
.3263ff2.png)
.0d83381.png)
.dec0422.png)




北京市医疗器械生产许可
北京市《医疗器械生产许可证》注销
从事医疗器械生产活动,应当遵守法律、法规、规章、强制性标准和医疗器械生产质量管理规范,保证医疗器械生产全过程信息真实、准确、完整和可追溯。
医疗器械注册人、备案人对上市医疗器械的安全、有效负责。
根据医疗器械风险程度,医疗器械生产实施分类管理。
从事第二类、第三类医疗器械生产活动,应当经所在地省、自治区、直辖市药品监督管理部门批准,依法取得医疗器械生产许可证;
从事第一类医疗器械生产活动,应当向所在地设区的市级负责药品监督管理的部门办理医疗器械生产备案。







项目内容/
Project Content
办证的条件
Conditions required for certification
 服务流程
服务流程 官方收费
官方收费 基础法律
基础法律 时间分布
时间分布 明细费用
明细费用 相关服务
相关服务 常见问题
常见问题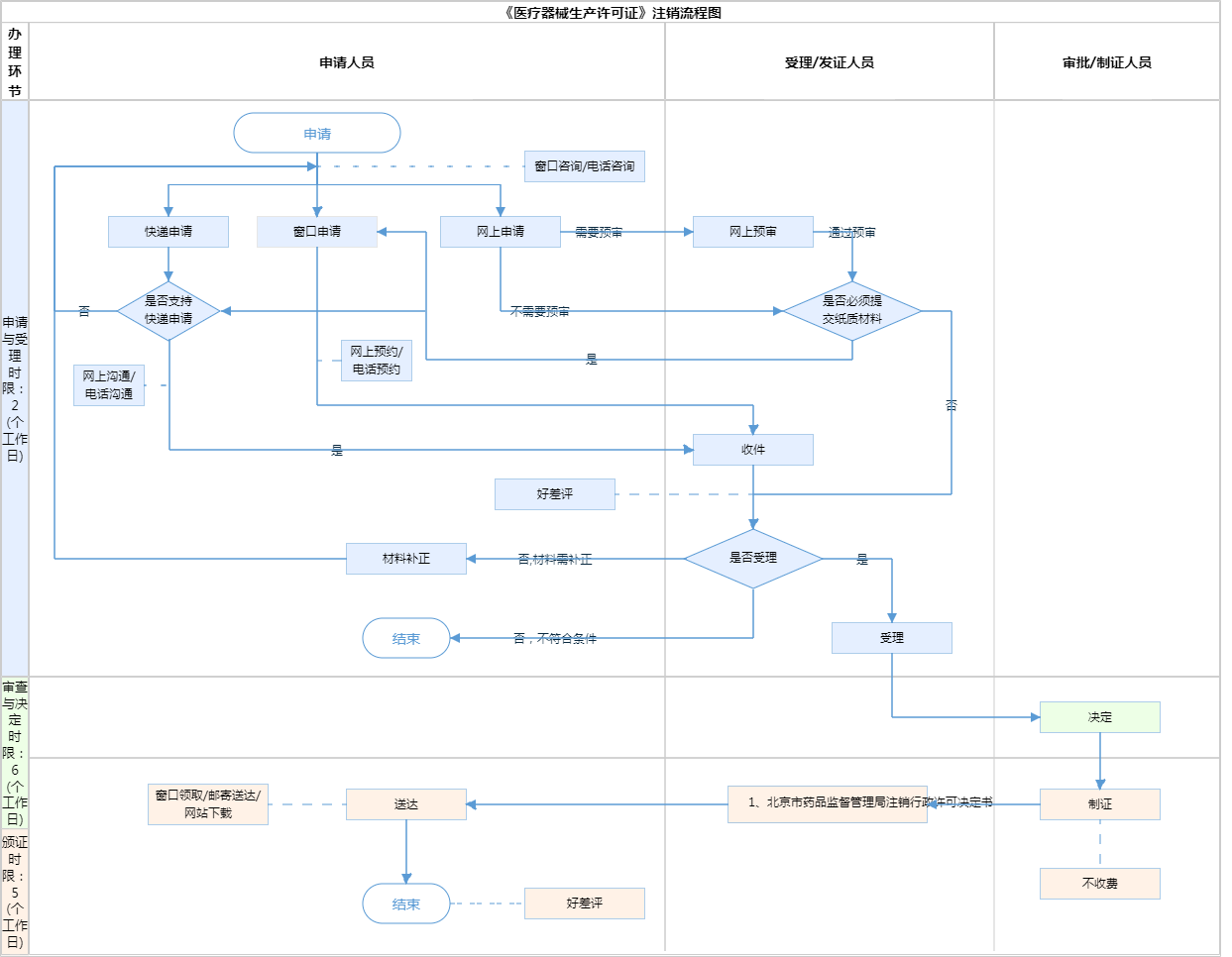
我们能提供的服务/
Services wecan provide
我们的优势/
Out davantage

专心敬业
我司拥有多位办理生产许可证15年的专家,拥有一批敬心专业的SC咨询辅导老师,能提供符合审查要求的车间以及 检验室规划设计;主导编制各类质量管理制度文件,设计质量记录表格,手把手指导记录填写。
经验丰富
我司有十五年的认证办理辅导服务经验,熟悉各类产品生产许可证审查细则、实施细则、工厂条件审查要求,熟悉每一个 环节的申办流程及注意事项,确保一次性审查通过。
关系网络优势
我司与全国各省质监局、食品药品监督管理局、 卫健委,辖区内地级市质监部门和食品药品监督管理部门建立了长 期良好的沟通渠道,人脉较广,顺利通过审查,办证速度快。
工程团队优势
有专业、经验丰富的厂房车间设计、工程装修、检验室规划、图纸绘制等方面的工程 技术人员,承接无尘车间,净化工程装修及改建工程,工程价格合理,省钱、省力、省心,确保硬件一次性通过。
资源需求便利优势
我司旗下有精密仪器,并与检测机构长期合作,检测价格优势,为企业在申请认证中提供各种方便。
人才输送优势
我司与专业技术院校长期合作,有一-批专兼职检验员提供, 可为企业顺利通过审查提供优越条件。





.36720e2.png)
在线咨询

陈老师

徐老师

王小姐

郭小姐

.2f44860.png)
热线电话
 18576401396 18575592846
18576401396 18575592846 .43b2c75.png)
转发网站
.4f4ea8a.png)
在线留言
.a3a1a2a.png)
回到顶部