
.3263ff2.png)
.0d83381.png)
.dec0422.png)




医疗器械相关新闻
About News
项目新闻
联系我们 400-690-0031 185-7559-2846
- 我们的服务
- 医疗器械音频广告审查
- 医疗器械图文广告审查
- 医疗器械视频广告审查
- 解决方案
.e60271e.png) 产品注册 产品注册注意事项想为。。。。。。undefinedundefined1,这是1点undefinedundefined2,这是两点undefinedundefined3,这是三点undefined
产品注册 产品注册注意事项想为。。。。。。undefinedundefined1,这是1点undefinedundefined2,这是两点undefinedundefined3,这是三点undefined.c944dfd.png) 产品认证 洁净室最主要之作用在于控制产品所接触之大气的洁净度日及温湿度,使产品能在一个良好之环境空间中生产、制造,此空间我们称之为洁净室。
产品认证 洁净室最主要之作用在于控制产品所接触之大气的洁净度日及温湿度,使产品能在一个良好之环境空间中生产、制造,此空间我们称之为洁净室。.5c9c52e.png) 产品检测 洁净室最主要之作用在于控制产品所接触之大气的洁净度日及温湿度,使产品能在一个良好之环境空间中生产、制造,此空间我们称之为洁净室。
产品检测 洁净室最主要之作用在于控制产品所接触之大气的洁净度日及温湿度,使产品能在一个良好之环境空间中生产、制造,此空间我们称之为洁净室。.ad5989a.png) 生产企业生产备案/许可 洁净室最主要之作用在于控制产品所接触之大气的洁净度日及温湿度,使产品能在一个良好之环境空间中生产、制造,此空间我们称之为洁净室。
生产企业生产备案/许可 洁净室最主要之作用在于控制产品所接触之大气的洁净度日及温湿度,使产品能在一个良好之环境空间中生产、制造,此空间我们称之为洁净室。

ANVISA注册咨询|I类产品提交产品技术文件存档以备随机审核2024-10-24
ANVISA认证认证审核相关申请资料,通过后,将在Diario Oficial da Uniao (DOU)上公布一个注册号,该注册有效期为5年。

巴西ANVISA注册咨询|为注册产品选择恰当的注册程序与管理类别有利于成功评估2024-10-24
ANVISA,正确的写法应是ANViSa,全称 Agência Nacional de Vigilancia Sanitária,隶属巴西卫生部,负责所有医疗器械、体外诊断产品及其它健康相关产品(如药品、卫生用品、化妆品等)的上市前审批与上市后监管。其角色相当于美国的FDA或中国的CFDA。 ANViSa制定的法规文件包括条例(RESOLUCAO DA DIRETORIA COLEGIADA,简称RDC)、指引(INSTRUCAO NORMATIVA,简称IN)及技术说明(NOTA TéCNICA),这些法规文件规定了有关产品及公司的注册和管理。值得注意的是,ANViSa的RDC和IN在修订时是重新编码的,并不像标准那样只改变年号而标准号固定,因此法规代号相同而年号不同时,多数情况下内容完全不同。比如RDC185:2001(医疗器械技术法规要求)和RDC185:2006(部分产品注册时需提供EIR);当然个别时候,法规代号相同而年号不同时,内容则是相关的,如IN7:2009和IN7:2010均是描述部分特殊I类和II类产品仍然需要进行Registro(注册)。这种做法,与中国CFDA的局令很相似,如不同年份颁布的5号令,内容可能完全不同,后颁布者通常并不是前一个年号的同一代号的文件的更新版。因此,学习和应用巴西法规文件时,应特别注意这一点。

TGA注册辅导|制造商需提供产品分类与GMDN代码及一致性声明文件后在ARTG登记2024-10-24
根据澳大利亚医疗用品法(Therapeutic Goods Act 1989)规定,所有在澳大利亚上市的医疗用品(药品和医疗器械)必须按有关要求,向澳大利亚医疗用品管理局(Therapeutic Goods Administration, TGA)提出注册或登记申请,获得注册登记(Australian Register of Therapeutic Goods,ARTG)后才能合法上市。

TGA注册辅导|登记成功后可在系统公开查询并合法在市场上销售2024-10-24
澳洲对医疗械准入和市场监管通过TGA下的两个部门分管,其中ODA(office of devices authorization)负责产品销售前的批准(对海外产品也就是准入),OPR(office of product review)负责产品投放市场之后的监督管理。

澳大利亚TGA注册辅导-对提交申请的文件进行全面评估保证产品符合基本原则2024-10-24
TGA负责监管澳大利亚的医疗设备和其他保健产品,例如细胞和组织,药品和血液制品。

TGA注册辅导|许可证有效维护作为制造商遵守澳大利亚法规及上市销售的关键2024-10-24
在澳大利亚,许可证维护是医疗器械制造商必须去认真对待的一个环节,如果不遵守规定带来的后果很可能就是失去市场准入、撤销批准、暂停商业活动和行政制裁罚款等一系列处罚,会严重影响到产品的销售。

澳大利亚TGA注册咨询|高风险产品将可能要求提供指定机构报告或生物学评估2024-10-24
TGA负责监管澳大利亚的医疗设备和其他保健产品,例如细胞和组织,药品和血液制品。

ANVISA注册辅导|企业文件必须符合ANVISA要求并有部分文件必须是葡萄牙语2024-10-23
ANViSa,全称Agência Nacional de Vigilância Sanitária,隶属巴西卫生部,负责所有医疗器械、体外诊断产品及其他健康相关产品(如药品、卫生用品、化妆品等)的上市前与上市后的管控。

ANVISA注册辅导|对成功注册的产品实施注册管理和认可制度并维护相关数据库2024-10-23
ANVISA认证通过对卫生医疗产品的生产和销售的管控,使之符合法律法规要求,从而促进和保护民众的健康。ANVISA认证对医疗器械产品实施注册管理和认可制度,并维护相关数据库。
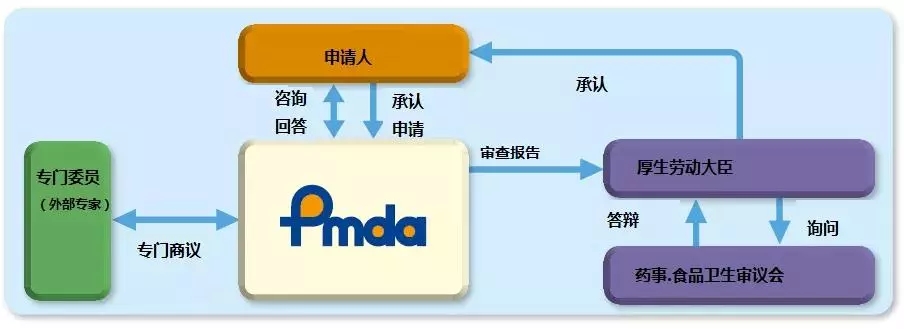
PMDA注册咨询|II类以上产品制造商取得QMS证书对于注册至关重要2024-10-23
对于在日本制造、进口和/或销售的医疗器械,日本医疗器械命名法(JMDN)代码和通用名称是参考ISO/TC210 GMDN项目中确定的医疗器械名称设置的。然后,通用名称根据其风险级别分为I类,II类,III类或IV类。这些分类是参照GHTF(全球协调工作队)的分类规则确定的。
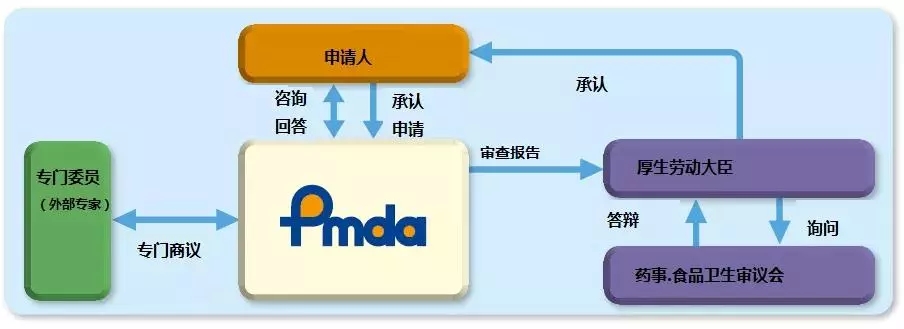
PMDA注册咨询|I类器械企业无需通过质量管理体系审核2024-10-23
对于在日本制造、进口和/或销售的医疗器械,日本医疗器械命名法(JMDN)代码和通用名称是参考ISO/TC210 GMDN项目中确定的医疗器械名称设置的。然后,通用名称根据其风险级别分为I类,II类,III类或IV类。这些分类是参照GHTF(全球协调工作队)的分类规则确定的。
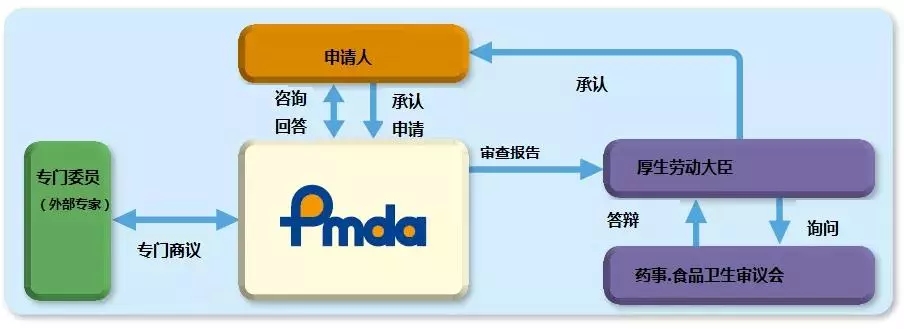
PMDA注册辅导|日本的医疗器械分类制度主要基于日本的医疗器械命名法规2024-10-23
从2014年11月25日起,日本《药事法》(亦称为PAL)经过修订,更名为《关于药品、医疗器械、再生细胞治疗产品、基因治疗产品和化妆品质量保证、功效和安全的法案》(简称PMD法)。PMD法提供了医疗器械、体外诊断试剂、处方药、药品和化妆品以及再生和细胞疗法产品在日本市场中的规管法律框架。PMD法的法律框架的管理和监督负责单位是日本卫生部,日本厚生劳动省(MHLW),制药和医疗器械局 (PMDA)日本的监管机构,与MHLW共同管理。该机构执行药品和医疗器械营销申请的科学评估,对其上市后的安全性进行监控。



.36720e2.png)
在线咨询

陈老师

徐老师

王小姐

郭小姐

.2f44860.png)
热线电话
 18576401396 18575592846
18576401396 18575592846 .43b2c75.png)
转发网站







.4f4ea8a.png)
在线留言
.a3a1a2a.png)
回到顶部